The Human Copy Number Variation community
Genomic variants are the basis of individual variations in human phenotypes and susceptibility to a large variety of diseases. Inherited variants can be the cause for syndromic or "rare" diseases, while neoplastic diseases ("cancer") arise from the accumulation of genomic variants in body tissues ("somatic mutations"), during the lifetime of an individual but sometimes associated with pre-disposing, inherited genome variations.
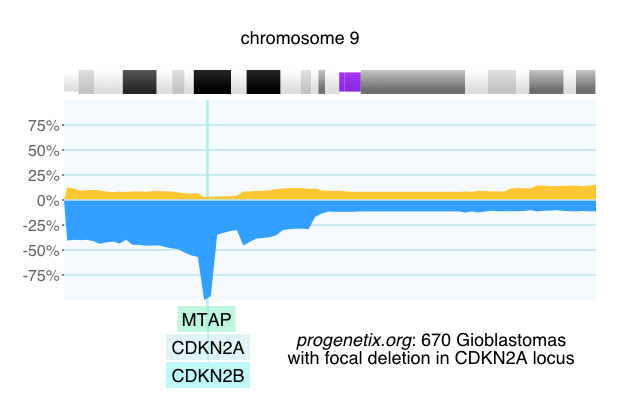 Among the different types of inherited and acquired genomic variants, regional genomic copy number variations (CNV) contribute - if measured by affected genomic sequences - contribute by far the largest amount of genomic changes, contributing both to many syndromic diseases as well as the vast majority of human cancers. These CNVs act through modifications of the dosage of genomic elements away from the base copy number, from the total abrogation of genetic function in homozygous deletions to genetic hyperactivity by copy number amlifications, sometimes resulting in tens or hundreds of copies of disease related genes.
Among the different types of inherited and acquired genomic variants, regional genomic copy number variations (CNV) contribute - if measured by affected genomic sequences - contribute by far the largest amount of genomic changes, contributing both to many syndromic diseases as well as the vast majority of human cancers. These CNVs act through modifications of the dosage of genomic elements away from the base copy number, from the total abrogation of genetic function in homozygous deletions to genetic hyperactivity by copy number amlifications, sometimes resulting in tens or hundreds of copies of disease related genes.
Despite the fact that Copy Number Variations are the most prevalent genetic mutation type, identifying and interpreting them still represents a major challenge. The ELIXIR human Copy Number Variation (hCNV) Community aims to implement processes to facilitate the detection, annotation and interpretation of these variations, and to engage scientists, clinicians and industry partners with significant involvement in areas relevant to the community.